U-M BME Researchers Create an 8D-Model of Cancer Metabolism
In a new study, researchers at the University of Michigan Department of Biomedical Engineering (U-M BME) have harnessed machine learning to successfully predict the metabolic inner workings of nearly 1,000 different cancer cell lines, creating a roadmap that could lead to highly tailored, synergistic combination therapies.
In the fight against cancer, understanding a cell’s metabolism—how it consumes nutrients and processes energy—is like discovering its fuel source. If scientists can map that fuel source, they can find ways to cut it off. However, a cancer cell’s metabolic engine is incredibly complex, regulated by an intricate web of genes, proteins, and cellular signals.
In a new study, researchers at the University of Michigan Department of Biomedical Engineering (U-M BME) have harnessed machine learning to untangle this complexity. The team successfully predicted the metabolic inner workings of nearly 1,000 different cancer cell lines, creating a roadmap that could lead to highly tailored, synergistic combination therapies.
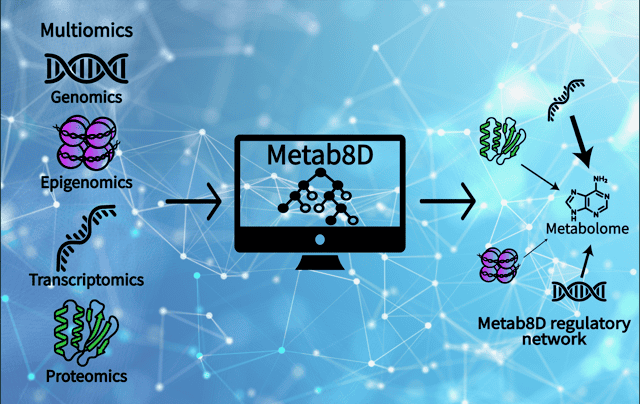
“People typically study these biological systems primarily using just one data set, such as transcriptomics or proteomics,” said Sriram Chandrasekaran, Associate Professor of Biomedical Engineering. “What we did here, which was very unique, is look at eight different data types—and up to 10 distinct sub-data sets all matched together—to see how they interconnect with metabolism. It gives us an eight-dimensional perspective on the cell.”
Decoding the Multiomic Maze
To understand how cancer cells control their metabolism, the U-M research team looked beyond just DNA. They utilized “multiomics”—an approach that examines multiple layers of biological data simultaneously.
The team fed their machine learning models data from eight distinct biomolecular classes across approximately 1,000 cancer cell lines. This massive dataset included everything from genetic mutations and structural DNA changes to transcriptomics (what genes are active), proteomics (the proteins actually produced from genes), and phosphoproteomics (how those proteins are turned on or off).
“We used the natural variation present across all these different cancer cells, where each cell line essentially has its own unique genetic barcode and unique metabolic state,” Dr. Chandrasekaran explained. “We then used machine learning to tease out how changes in one layer could be predictive of another layer.”
The analysis revealed that a cell’s overall metabolism is most tightly linked to its transcriptome (the RNA molecules in a cell), with both coding and non-coding RNAs emerging as the top predictors of metabolic behavior.
Central vs. Peripheral Metabolism: Two Different Controls
One of the study’s most surprising findings was that not all metabolic pathways are regulated the same way. The researchers discovered a clear divide between “peripheral” and “central” metabolism, which Dr. Chandrasekaran explains using a city traffic analogy.
“Imagine a city where each metabolite is a traffic intersection,” said Dr. Chandrasekaran. “We found some metabolites that are regulated at a local level on the periphery of the system. If you just know what is coming in and going out of that intersection, you can predict the metabolite levels. But at intersections in the middle of the city, i.e., central metabolism, regulation is extremely complex. You need to know almost the entire city’s traffic map to predict its levels.”
Peripheral Metabolism: These are the specialized, outer-edge chemical pathways in a cell. The AI found that these processes are relatively straightforward to predict because the level of a specific metabolite directly matches the level of the enzyme responsible for making it.
Central Metabolism: This is the core engine room of the cell, responsible for essential functions such as generating energy and maintaining cellular health (redox balance). Here, the machine learning model found that looking at enzymes alone wasn’t enough. Central metabolism requires a complex interplay among overarching cell-signaling and redox pathways and often does not reflect the genes expressed in that specific pathway, as is commonly assumed by scientists.
Finding Cancer’s Vulnerabilities
By tracing these complex interactions, the AI reconstructed detailed networks showing exactly how highly predictable metabolites are regulated. Through this process, a specific cell signaling pathway, known as YAP1, emerged as a global mastermind, regulating cancer metabolism across four distinct biological layers.
Crucially, the U-M team didn’t just map these networks; they used the AI to find vulnerabilities to design smarter drug combinations.
“Cancer cells are highly resilient; if you block a certain gene, they figure out a way to evade the drug attack and grow,” Dr. Chandrasekaran said. “Using our traffic analogy, if there are two parallel roads that go to the same destination and you only block one, traffic will just reroute. But if you block both routes, there is no way to reach the destination.”
By identifying these “metabolic back-up generators,” the research provides a data-driven strategy for identifying what scientists call synthetic-lethal interactions—pairs of targets that are harmless when blocked individually, but deadly to the cancer cell when disrupted together. The team found that drugs designed to block these complementary routes were statistically far more synergistic than random drug pairings.
An Interdisciplinary, Student-Led Effort
The entire predictive model, named Metab8D, has been made available online as an open-access resource for the broader scientific community. “It works like a multilayer Google Maps for cellular metabolism,” Dr. Chandrasekaran noted. “You can zoom in or zoom out. If a researcher wants to know how a specific metabolite like citric acid is predicted, they can look at each step the model used and see the top features.”
The massive undertaking required extensive data curation, which became a hallmark of student mentorship and interdisciplinary collaboration at U-M BME. The project was spearheaded by Ryan Schildcrout, a graduate student whose entire academic journey has evolved alongside the research.
“Ryan started this project as an undergraduate, continued working on it as a master’s student, and is now a Ph.D. student, finally seeing his work published,” said Dr. Chandrasekaran. “He led an army of undergraduate students from chemical engineering, biochemistry, and biomedical engineering to carry out this data collection. It was a truly interdisciplinary student team that made this biomedical application possible.”
“With all the biological data that exist, it’s important to build tools to rigorously and effectively explore them,” Schildcrout said. “Dr. Chandrasekaran’s mentorship has been invaluable in preparing me to contribute to cancer research–especially for studying modes of regulation we know less about, like non-coding RNAs. My time in Dr. Chandrasekaran’s lab has shown me the breadth and depth of what computational research can teach us. I’m excited to use those lessons to continue to piece together the inner workings of cancer metabolism.”
This research was spearheaded by the laboratory of Sriram Chandrasekaran, Associate Professor of Biomedical Engineering at the University of Michigan, alongside lead researchers including graduate student Ryan Schildcrout. The work was supported by faculty start-up funds from the University of Michigan, the Camille and Henry Dreyfus Foundation, the Rogel Cancer Center at UM, the National Institutes of Health (NIH) National Institute of General Medical Sciences, the Advanced Proteogenomics of Cancer Program, and the Graduate Assistance in Areas of National Need (GAANN) Fellowship.
Reference:
Metab8D: a metabolic regulome network from multiomics and machine learning